








{kind=link}
{kind=link}
{kind=link}
INTRODUCTION
Keystone virus (KEYV) is a member of the genus Orthobunyavirus and is the sole member of the species Keystone orthobunyavirus.1 It was first identified in 1964 in Keystone, a small town in west-central Florida near Tampa.2 Serologic studies after its identification suggested that approximately 20% of the population in endemic areas had antibodies to the virus3; there has been at least one case of meningoencephalitis in a child in which KEYV was implicated as a possible etiologic agent,4 and it recently was isolated from a teenager in north-central Florida with a fever/rash syndrome.5 KEYV was initially placed within what was designated as the California encephalitis serogroup, which included other Orthobunyavirus spp. that were associated with meningoencephalitis in humans, such as La Crosse virus and Jamestown Canyon virus (JCV). It is noteworthy that the illnesses they cause are currently on the U.S. CDC list of notifiable diseases. Although the California encephalitis serogroup classification scheme is no longer used, the association with other viruses linked to meningoencephalitis raises questions about whether KEYV might be an underrecognized cause of illness (including meningoencephalitis) among persons living in the southeastern United States, particularly as identification of the virus in clinical cases has not been possible outside of a research setting.
Aedes atlanticus has been identified as the primary vector for KEYV in prior studies, and there are data suggesting that the virus can undergo transovarian transmission in this species.6–12 The virus has also been associated with 10 other types of mosquitoes, including Culex spp., but the role of these other vector species in KEYV transmission and infection remains uncertain.8,9,13 Limited data are available on vertebrate hosts that sustain the virus in a sylvatic cycle, although cotton rats (Sigmodon hispidus) and rabbits (Sylvilagus floridanus) are known to be amplifying hosts; there are no data to assess the possibility of an urban transmission cycle in which humans might play a role. Despite the fact that KEYV has been recognized for more than 50 years, sequence data are available for only four KEYV genomes from outside of our group (Table 1), which has limited the ability of investigators to assess strain diversity and evolutionary trends. To better characterize the dynamics of KEYV in mosquito populations (and as an initial step in understanding the risk of KEYV infection in humans), we screened mosquito pools from St. Johns County on Florida’s east coast for KEYV by real-time reverse transcriptase polymerase chain reaction (rRT-PCR), virus isolation in cell cultures, genomic sequencing of virus isolates, and PCR of mosquito DNA for verification of mosquito species.
KEYV strains for which sequence data are available
| Strain | Host | Origin | Collection date | Segment L NCBI accession no. | Segment M NCBI accession no. | Segment S NCBI accession no. |
|---|---|---|---|---|---|---|
| AR14033 | Culex sp. | TX | May 2016 | MG821229.1 | MG821230.1 | MG821231.1 |
| AVA1709441 | Culex sp. | Orange County, TX | Aug. 2017 | MG765469.1 | MG765470.1 | MG765471.1 |
| B64-5587.05 | Aedes cf. atlanticus/ tormentor | FL | 1964 | NC_043629.1 | NC_043627.1 | NC_043628.1 |
| KEYVLK01 | Ae. atlanticus | Sarasota County, Fl | June 2005 | KT630288.1 | KT630289.1 | KT630290.1 |
| KEYVLK02 | Ae. atlanticus | Sarasota County, FL | June 2005 | KT630291.1 | KT630292.1 | KT630293.1 |
| Gainesville-1/2016 | Homo sapiens | Alachua County, FL | Aug. 2016 | MH016784.1 | MH016785.1 | MH016786.1 |
| St. Johns County-FL-1/2019 | Ae. atlanticus | St. Johns County, FL | July 2019 | MT127621.1 | MT127622.1 | MT127623.1 |
| St. Johns County-FL-2/2019 | Ae. atlanticus | St. Johns County, FL | Oct. 2019 | MZ156786.1 | MZ156785.1 | MZ156784.1 |
| St. Johns County-FL-3/2019 | Ae. atlanticus | St. Johns County, FL | Oct. 2019 | MZ156789.1 | MZ156788.1 | MZ156787.1 |
| St. Johns County-FL-4/2019 | Ae. atlanticus | St. Johns County, FL | Oct. 2019 | MZ156792.1 | MZ156791.1 | MZ156790.1 |
| St. Johns County-FL-5/2019 | Ae. atlanticus | St. Johns County, FL | Oct. 2019 | MZ156795.1 | MZ156794.1 | MZ156793.1 |
KEYV = Keystone orthobunyavirus; NCBI = National Center for Biotechnology Information; no. = number.
MATERIALS AND METHODS
Sampling sites and strategy.
The Anastasia Mosquito Control District (AMCD) is responsible for mosquito surveillance and control in St. Johns County, Florida, which includes the city of St. Augustine. Routine surveillance is conducted by using CDC light traps (John W. Hock, Gainesville, FL) baited with CO2 and placed along with BG-Sentinel traps (BioGents, Regensburg, Germany), baited with BG lure, for mosquito capture. For the current study, mosquitoes in traps were anaesthetized via exposure to CO2 gas until no movement was observed. Taxonomists at AMCD identified mosquitoes to the species level by using taxonomic keys,14 and mosquitoes were then separated into pools by species, sex, date of collection, and trap type and location. For the current study, mosquito pools were stored at −80°C pending transfer to the arbovirus research laboratory at UF/EPI for further analyses. To confirm mosquito species identification in a subset of Ae. atlanticus mosquito pools, PCR was used to amplify ∼710 bp of the mitochondrial cytochrome C oxidase subunit I (COI) gene from mosquito pool samples. The amplified segments were then sequenced, and the results subjected to nucleotide Basic Local Alignment Search Tool (National Center for Biotechnology Information) analyses to confirm identity as previously described at Aix Marseille Université in France.15,16
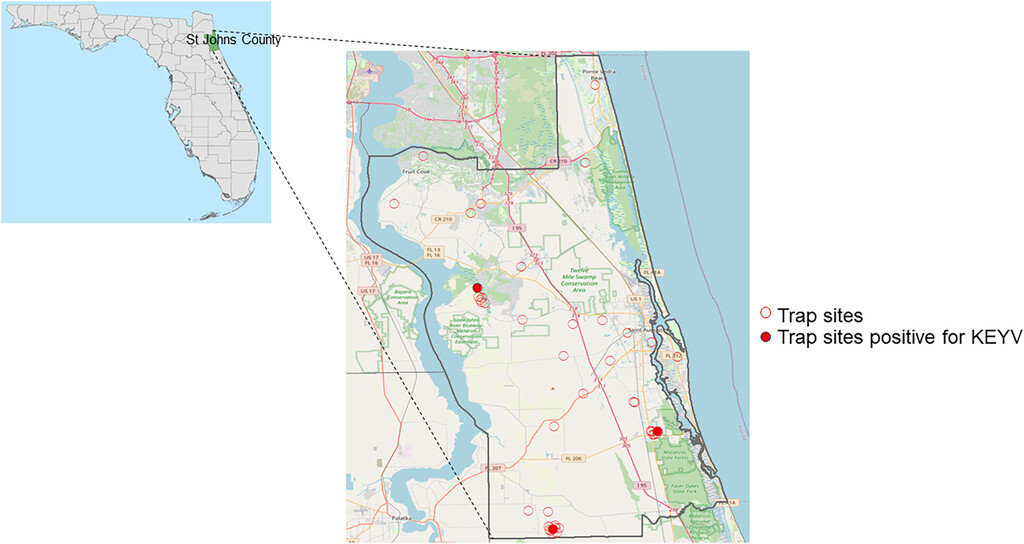
Samples were collected from 43 sites in St. Johns County (Figure 1). Mosquito pools that contained Ae. atlanticus, Aedes albopictus, and Aedes infirmatus were screened using a multiplex RT-PCR for KEYV and Melao virus (MELV) as described subsequently. In 2020, additional sampling sites were selected around each of the positive sites from 2019, and screening at these sites was expanded to include collection and screening of mosquito pools containing the most common Culex species in northeast Florida (Cx. coronator, Cx. melanoconian, Cx. quinquefasciatus and Cx. salinarius).

Map of sampling locations in St. Johns County. Map showing St. Johns County within Florida (left), and mosquito trapping sites in St. Johns County (red circles), showing the three trapping sites (filled red circles) for which mosquito pools were found positive for Keystone orthobunyavirus (KEYV) (right). County boundaries derived from the U.S. Census Bureau: American Community Survey 2010, from the Florida Geographic Data Library (FGDL, fgdl.org, accessed June 6, 2022). Basemap data for St. Johns county were obtained from Open Street Map (OSM), © OpenStreetMap (and) contributors, CC-BY-SA (openstreetmap.org).
Citation: The American Journal of Tropical Medicine and Hygiene 108, 6; 10.4269/ajtmh.22-0594
Real-time RT-PCR.
A novel KEYV/MELV primer and probe mix was designed at Aix Marseille Université in France to detect the genomic RNA of Orthobunyavirus spp. KEYV and MELV,17 which are closely related genetically, as well as other viruses, such as Serrado Navio, Inkoo, Jameston Canyon, Jerry Slough, and South River. Alignments are presented as supplemental material (Supplemental Document 1). Sequences of the primers and probes are presented in Table 2; the probes were 5′ labeled with the same dye (6-FAM). A synthetic standard RNA was used for assessing analytical sensitivity. The target regions, included in a plasmid synthetized by Genscript (GenScript, Piscataway, NJ), were amplified by PCR. The RNA transcript was synthetized in vitro using the MEGAshortscriptTM T7 Transcription Kit (Invitrogen-Thermo Fisher ScientificTM, Waltham, MA), and the RNA concentration was determined using a Thermo Scientific NanoDropTM (Thermo Fisher Scientific). The RNA transcript was serially diluted from 108 to 102 copies/µL, and dilutions were stored at –80°C. Limit of detection (LoD95) of the lyophilized reagents was calculated as previously described.18–20 Briefly, the evaluation of the sensitivity was done by using 14 serial dilutions of the quantified in vitro transcribed RNA containing 3.45 × 103 to 4.21 × 101 RNA copies/µL tested using 12 replicates for each. A Ct value > 40 was considered negative. The lower LoD was determined by probit regression analysis, using IBM SPSS Statistics software version 24. The LoD95 was defined as a concentration of viral copies, achieving a 95% hit rate. LoD95 was 104.93 (95% CI: 62.97–257.17; Supplemental Document 2).
KEYV small genome rRT-PCR primers and probes
| Primer probe name | Sequence | Conc in Rx |
|---|---|---|
| MELcxS35-56 (F) | CAGGTGCAAATGGATTTGATCC | 400 nM |
| MELcxP113-137 (P) | CCGTTAGGATCTTYTTCCTTAATGC | 160 nM |
| MELCXR164-145 (R) | CGAGHGAGAGCAGYTTTGGC | 400 nM |
| MELcxS193-212 (F) | TTTGGAGASTGGCAGGTGGA | 400 nM |
| MELCXP218-224 (P) | TCAAYAATCATTTTCCTGGRAACAGGA | 160 nM |
| MELCXR273-249 (R) | TRAGATCGTTGTTACCAATTGGG | 400 nM |
Conc in Rx = concentration in reaction; F = forward primer; KEYV = Keystone orthobunyavirus; P = probe; R = reverse primer; rRT-PCR = real-time reverse transcriptase polymerase chain reaction.
Mosquito pools containing no more than 25 mosquitoes per pool were homogenized in chilled phosphate-buffered saline (PBS) and a mixture of 2 mm and 0.1 mm high-density yttrium-zirconium beads (Glen Mills, Clifton, NJ) that had been chilled at –20°C.21 Briefly, homogenates were prepared by bead-beading mosquitoes in 750 µL of PBS (Gibco, Thermo Fisher Scientific, Waltham, MA) in a Bead-Bug (Benchmark Scientific Sayerville, NJ) at 3,000 vibrations/minute for 90seconds, and the tubes were immediately moved to a chilled rack. The tubes were then centrifuged at 8,000 rpm for 1 minute to pellet the mosquito debris, and 140 µL of the supernatant was immediately added to 560 µL of Qiagen QIAamp virus RNA Extraction Buffer (Valencia, CA) for RNA extraction using the QIAamp viral RNA extraction kit according to the manufacturer’s instructions. The remaining ∼500 µL of the supernatant was transferred into sterile tubes containing 500 µL filtered 20% w/v trehalose in PBS to attain a final concentration of 10% (w/v) trehalose and stored immediately at –80°C until further analyses.
Real-time reverse transcriptase polymerase chain reaction was carried out using Super ScriptTM III One Step RT-PCR with Platinum Taq polymerase (Thermo Fisher Scientific) as follows: 15 minutes at 50°C for the RT step, 2 minutes for Taq activation at 95°C, then 45 cycles of 15 seconds for denaturing at 95°C followed by 45 seconds for annealing and extension at 60°C.
Cell lines.
Two cell lines that are susceptible and permissive for KEYV were obtained from the American Type Culture Collection (ATCC, Manassas, VA): C6/36 (Aedes albopictus (mosquito), ATCC CRL1660) and Vero E6 (Cercopithecus aethiops (African green monkey) kidney, ATCC CRL 1586). Both cell lines were grown as monolayers in a humidified atmosphere containing 5% CO2, the C6/36 cells at 28°C and Vero E6 cells at 37°C as described by Ahasan et al.22
Virus isolation.
Mosquito pools with rRT-PCR Cq values < 38 were considered positive, and virus isolation was attempted in C6/36 and Vero E6 cells. In brief, 100-uL aliquots of unfiltered mosquito-pool supernatant were inoculated onto 2.8 × 106 C6/36 cells and nearly confluent monolayers of Vero E6 cells in filter-cap T-25 flasks (Corning, Corning, NY), which were then incubated in 28°C and 37°C incubators, respectively, in the presence of 5% CO2. The cells were refed with cell culture maintenance media consisting of advanced Dulbecco’s modified Eagle medium (aDMEM) supplemented with gamma-irradiated, low-antibody, heat inactivated, 3% fetal bovine serum (HyClone, Logan, UT), penicillin-streptomycin-neomycin (Gibco, Thermo Fisher Scientific), and Glutamax-1 supplement (Gibco; at a final concentration of 3 mM). The cell maintenance media was changed every 3 days. The infected Vero E6 cells were observed for 14 days for any visible virus-induced cytopathic effects (CPE). The cell maintenance media of the C6/36 cell lines was blindly tested by rRT-PCR once every 3 days for virus because KEYV CPE may be difficult to detect in those cells. Vero-E6 cells that displayed CPE were scraped off the growing surface of the flasks and the cells and the maintenance media were tested by rRT-PCR for KEYV (or a closely related member of the California encephalitis serogroup viruses).
Next-generation sequencing of virus genomes.
Next-generation sequencing was attempted on virus RNA purified from cell culture media wherein a Cq value of < 20 was attained in rRT-PCR tests. cDNA libraries were prepared using the NEB Ultra II RNAseq Library Prep Kit (New England Biolabs, Ipswich, MA), and sequencing was performed using a version 3 chemistry 600-cycle kit that was run on a MiSeq sequencer (Illumina, San Diego, CA). Host (i.e., African Green Monkey for Vero E6 cells) reads were removed using Kraken v2.0. De novo assembly was performed in CLC Genomics workbench v.10.
Phylogenetic analyses.
Whole genome sequence alignments were performed with MAFFT23 and manually refined on AliView.24 Recombination and reassortment analyses were performed with an algorithm based on the PHI test25,26 implemented SplitsTree527 and with RDP4.28 Presence of phylogenetic signal was verified by Likelihood mapping,29 as implemented in IQTREE v.2.0.6.30 Maximum likelihood trees were calculated with the same IQTREE version, with the best fitting nucleotide substitution model according to the Bayesian information criterion and 1,000 bootstrap replicates. Nucleotide and amino acid p-distances were calculated with Mega-X v.10.0.3.31 Trees were visualized and exported as image using FigTree v.1.4.4.32
Plaque assay.
Plaque assays were performed in Vero E6 cells using a standard agarose overlay method as described by Hamilton et al.33 Briefly, newly confluent Vero E6 cells grown in six-well cell culture plates were inoculated with 0.2 mL of virus serially diluted in aDMEM. The virus was adsorbed to the cells for 1 hour at 37°C with manual rocking of the plates performed every 15 minutes. After virus adsorption, the cells were washed with aDMEM and the wells overlaid with 3 mL/well of primary overlay consisting of 1.6% w/v agarose (Invitrogen) mixed 1:1 with 2× complete Eagles’ modified essential medium (EMEM; Lonza, Walkersville, MD) containing 20% fetal bovine serum and antibiotics. The plates were inverted and incubated for 3 days at 37°C, then overlaid with 2 mL of secondary overlay of 1.6% w/v agarose mixed 1:1 with 2× EMEM containing serum, antibiotics, and 0.14 mg/ml neutral red (catalog no. N2889; Sigma-Aldrich, St. Louis, MO), the plates inverted, and incubated for 2 additional days to visualize plaques.
RESULTS
As shown in Table 3, 293 mosquito pools (total of 2,171 mosquitoes) met our criteria for screening by the KEYV/MELV rRT-PCR. Criteria included being in a pool with Ae. atlanticus, Ae. infirmatus, or Ae. albopictus or in a Culex pool collected in a location where rRT-PCR-positive pools had been identified in 2019. Of the 98 Ae. atlanticus pools tested, 10 were positive by the KEYV/MELV rRT-PCR test. Multiple positive pools were collected at three sites (three to four positive pools/site), as highlighted in Figure 1; all positive pools were collected in Fall 2019. Because of uncertainties about differentiation of Ae. atlanticus and Ae. informatus in four of the 10 KEYV/MELV rRT-PCR positive pools, COI amplicons were sequenced to confirm mosquito identity; in all instances, the sequenced COI amplicons had 99% identity with that of Ae. atlanticus (GenBank JX259518.1) with no evidence of similarity with Ae. infirmatus. Of the 211 Culex spp. pools tested, 10 pools containing either Cx. coronator or Cx. salinarius were positive with the KEYV/MELV rRT-PCR.
Number of mosquitoes/mosquito pools tested with KEYV/MELV rRT-PCR, by species and year, June 2019–April 2020
| Species | 2019 | 2020 | ||||
|---|---|---|---|---|---|---|
| Total no. of tested mosquitoes | No. of tested pools | No. of positive pools | Total no. of tested mosquitoes | No. of tested pools | No. of positive pools | |
| Aedes albopictus | 569 | 62 | 0 | 0 | 0 | 0 |
| Aedes atlanticus | 518 | 68 | 10 | 49 | 30 | 0 |
| Aedes infirmatus | 129 | 40 | 0 | 691 | 65 | 0 |
| Aedes spp (unidentified) | 166 | 28 | 0 | 0 | 0 | 0 |
| Culex coronator | 0 | 0 | 0 | 49 | 19 | 9 |
| Culex salinarius | 0 | 0 | 0 | 344 | 87 | 1 |
| Culex quinquefasciatus | 0 | 0 | 0 | 35 | 20 | 0 |
| Culex melanoconion | 0 | 0 | 0 | 231 | 53 | 0 |
KEYV = Keystone orthobunyavirus; MELV = Melao virus; No. = number; rRT-PCR = real-time reverse transcriptase polymerase chain reaction.
Homogenates from two of the 10 KEYV/MELV rRT-PCR-positive pools of Ae. atlanticus induced CPE after inoculation onto Vero-E6 cells but did not produce readily discernable CPE in C6/36 cells. A nearly complete genome sequence was obtained for all three KEYV genome segments from spent culture medium from one of these two rRT-PCR-positive mosquito pools (virus isolate: St. Johns County-FL-1/2019; details and accession numbers are provided in Table 1). When sequencing was attempted on spent cell culture medium from the second Vero E6 cell culture showing CPE, there were unreadable sections across the L, M, and S genome segments, consistent with the occurrence of nucleotide polymorphisms indicating the presence of more than one virus variant in the sample. In a plaque assay33 of the second KEYV positive batch, five well-separated plaques were picked and inoculated onto fresh Vero E6 and C6/36 cells. Four of the resulting virus isolates were successfully resequenced by next-generation sequencing, revealing the presence of three distinct KEYV variants: St. Johns County-FL-3/2019; St. Johns County-FL-4/2019; and isolates St. Johns County-FL-2/2019 and St. Johns County-FL-5/2019, which were identical. No virus was detected in the other plaque from Vero E6 cells.
A third KEYV/MELV rRT-PCR-positive mosquito pool of Ae. atlanticus induced CPE in C6/36 cells but not in Vero-E6 cells. On sequence analysis of C6/36 culture media Long Pine Key virus, a flavivirus, was identified; the sequence of this isolate has subsequently been posted in GenBank (MZ090957.1). It is unclear whether this flavivirus generated a positive rRT-PCR signal using the KEYV primer detection assay or if there was a yet unidentified orthobunyavirus that may have been present in the mosquito pool but was not recovered in cell culture.
None of the 10 KEYV/MELV rRT-PCR-positive pools from Culex spp. produced CPE in cell culture, and efforts at virus isolation were unsuccessful. For rRT-PCR-positive pools from Culex spp. and Ae. atlanticus from which virus isolation was not possible, samples had an average Cq value of 32.
Phylogenetic analyses.
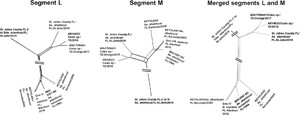
Three separate alignments were generated, one for each genomic segment (designated as L, M, and S), using all available KEYV sequences from the National Center for Biotechnology Information (NCBI) database and sequence data from the current study (Table 1). The likelihood mapping analyses showing the segment L and M alignments have a strong phylogenetic signal (Figure 2A and B) and therefore could be used for phylogenetic inference; in contrast, the segment S alignment has a much weaker signal (Figure 2C). No evidence of within-segment recombination was found in any of the segments.
Maximum likelihood (ML) trees of Keystone orthobunyavirus (KEYV) strains. ML trees were inferred from 11 genome sequences, using the best fitting nucleotide substitution models as detected by Bayesian information criterion. Branches are scaled in number of nucleotide substitutions per site according to the bar below each tree. Nonparametric bootstrap values (1,000 replicates) are indicated along supported branches. The two clades hosting St. Johns county isolates are colored in red and blue. On the side of each tree, likelihood mapping shows the amount of phylogenetic signal in each alignment; details on this analysis are given in the methods section. Briefly, the higher the sum of the percentages in the triangle corners (completely resolved quartets), the higher is the phylogenetic signal contained in the alignment. The alignment used for the S segment in panel (C) has low phylogenetic signal (resolved quartets < 60%), and therefore caution must be used in drawing conclusions on the base of this tree results; still, the segment S tree topology matches the one for segment L, which has strong phylogenetic signal and bootstrap support. The tree inferred from segment M has a different topology from the other segments, a clear indication that genome rearrangement has occurred in the ancestry of the sequences. (A) Segment L ML tree; (B) segment M ML tree; and (C) segment S ML tree.
Citation: The American Journal of Tropical Medicine and Hygiene 108, 6; 10.4269/ajtmh.22-0594
Maximum likelihood (ML) trees, calculated with IQTREE v.2.0.6 (36), showed different phylogenetic histories, depending on the segment. An ML tree from segment L (Figure 2A) showed the St. Johns isolates, all obtained from Ae. atlanticus mosquitoes, clustering with KEYV sequences from Culex spp. from Texas. The other Florida KEYV sequences were derived from Ae. atlanticus and a human, respectively in Sarasota and Alachua counties, and clustered with the reference strain originally isolated in 1964. These two clades shared 86.4% nucleotide identity, and 96.9% amino acid identity in segment L. The segment M tree (Figure 2B) showed the St. Johns isolates from October 2019 grouped separately from the July isolate and all other sequences. Segment S alignment, although afflicted with low phylogenetic signal, matched segment L tree topology. The trees show two major clades, with high bootstrap support and high divergence.
From the tree topologies, it appears that these viruses underwent reassortment events. To explore this hypothesis, we concatenated the L and M segments; we then applied algorithms that are normally used to test for recombination to assess such pseudo-sequences. At a first analysis by neighbor net and phi test (Figure 3), the new alignment showed strong evidence for reassortment (P < 10−6). RDP4 algorithms28 confirmed these results, recognizing the correct breakpoint coordinates within 20 nucleotides (Supplemental Document 3). The program results also corroborated the existence of unknown parental strains to the reassorted genomes.
Neighbor net plots of segments L and M and merged sequences. Neighbor net was inferred from pair-wise p-distances of the 11 genomes, for segments L and M, separately, and then from the alignment resulting from merging the two fragments for each genome. Within-segment recombination for L and M, and rearrangement for the merged sequence was assessed by the PHI test. No significant evidence of recombination was found in either segment (PHI test P > 0.001), while a significant P value was found for the merged alignment (P < 10−6).
Citation: The American Journal of Tropical Medicine and Hygiene 108, 6; 10.4269/ajtmh.22-0594
DISCUSSION
Keystone orthobunyavirus is, in many ways, a “forgotten” virus. First identified in 1964,2 with a series of subsequent studies documenting its presence at multiple environmental sites in the south and southeastern United States,6–12 there are few data available on the role of KEYV as a human pathogen, due in large part to the lack of a readily available test for clinical infections. Nonetheless, human infections with KEYV would appear to be common in the region, with studies in the 1960s and 1970s reporting that 19% to 21% of serum samples from normal, healthy people in the Tampa Bay region had neutralizing antibodies against the virus.3 In keeping with these findings, we recently screened 31 random, deidentified serum samples from the laboratory at UFHealth/Shands Hospital and found that nine (29%) of the 31 samples had neutralizing antibodies to KEYV, with titers as high as 1:40 (personal communication, J. Lednicky). This is comparable to the 28% seroprevalence seen with JCV (currently the third leading cause of arboviral neuroinvasive disease in the United States) in Michigan and the upper Midwest.34 Of note, since the implementation of IgM assays for JCV at the CDC in 2013, there has been a striking increase in the number of neuroinvasive JCV cases identified nationally, including first-time reports of cases from eight states,35 highlighting the critical importance of having diagnostic tests available to identify infections and recognize associated human illness.
The current study documented the continued circulation of KEYV in regional mosquito populations in Florida, with the virus being isolated from Ae. atlanticus mosquito pools. However, further studies are needed to assess the sensitivity of the rRT-PCR assay used in this study, as well as its specificity for other Orthobunyavirus species; nonetheless, its incorporation into an easily usable assay kit, as has been done by Aix Marseille Université, is a promising start toward development of human diagnostics. The significance of the positive rRT-PCR assay results obtained in the current study with Culex spp. is uncertain. In studies conducted by the CDC, KEYV has been isolated and sequenced from Culex species.13 Our failure to isolate the virus from Culex may reflect issues surrounding viability of the virus in Culex and/or viral load. It is also possible that the screening assay was picking up other cross-reacting viruses that were not amenable to our culture techniques.
Our genome sequence data are somewhat difficult to interpret, due in part to the small number of available KEYV sequences. Genetic analysis of the L segment showed evidence of two widely separated clades (86.4% nucleotide identity and 96.97% amino acid identity). The International Committee on Taxonomy of viruses requires less than 96% identity in the complete amino acid sequence of the L segment to define different species within the genus Orthobunyavirus.1 Although our clades did not quite meet these criteria, the degree of diversity is substantial, and, with additional data, it may be necessary to separate out species or subspecies of KEYV. Further, subsequent analyses suggested the possibility of earlier reassortment events with currently unidentified progenitor strains. Taxonomic classification might need to include whole genome analyses to detect the divergence and appearance of new strains/species after such reassortment events. Clearly, further studies (with sequencing of additional genomes) are needed to sort out underlying evolutionary pathways and possible associations between specific strains and geographic location, host species, and, possibly, clinical outcome.
ACKNOWLEDGMENTS
We thank the staff at AMCD for their assistance in sample collection and initial analysis, with particular thanks to Heather Ward, who was involved with mosquito identification and pooling of samples, and Mandy Pearson. We also thank Drs. Thomas B. Waltzek and Kuttichantran Subramaniam for assistance with next-generation sequencing and Dr. Laurence Thirion and Pierre Combe for valuable help in assay qualification.
REFERENCES
- 1.↑
Abudurexiti A et al., 2019. Taxonomy of the order Bunyavirales: update 2019. Arch Virol 164: 1949–1965.
- 2.↑
Bond JO , Hammon WM , Lewis AL , Sather GE , Taylor DJ , 1966. California group arboviruses in Florida and report of a new strain, Keystone virus. Public Health Rep 81: 607–613.
- 3.↑
Parkin WE , Hammon WM , Sather GE , 1972. Review of current epidemiologic literature on viruses of the California arbovirus group. Am J Trop Med Hyg 21: 964–978.
- 4.↑
Gates EH , Bond JO , Lewis AL , 1968. California group arbovirus encephalitis in Florida children. J Fla Med Assoc 55: 37–40.
- 5.↑
Lednicky JA , White SK , Stephenson CJ , Cherabuddi K , Loeb JC , Moussatche N , Lednicky A , Morris JG Jr. , 2019. Keystone virus isolated from a Florida teenager with rash and subjective fever: another endemic arbovirus in the southeastern United States? Clin Infect Dis 68: 143–145.
- 6.↑
Watts DM , Bailey CL , Roberts NT , Ammariello RF , Dalrymple JM , Clark GC , 1988. Maintenance and transmission of Keystone virus by Aedes atlanticus (Diptera: Culicidae) and the gray squirrel in the Pocomoke Cypress Swamp, Maryland. J Med Entomol 25: 493–500.
- 7.↑
Roberts DR , Scanlon JE , 1975. The ecology and behavior of Aedes atlanticus D. & K. and other species with reference to Keystone virus in the Houston area, Texas. J Med Entomol 12: 537–546.
- 8.↑
LeDuc JW , Burger JF , Eldridge BF , Russell PK , 1975. Ecology of Keystone virus, a transovarially maintained arbovirus. Ann N Y Acad Sci 266: 144–151.
- 9.↑
LeDuc JW , 1978. Natural transmission of Keystone virus to sentinel rabbits on the Delmarva Peninsula. Am J Trop Med Hyg 27: 1041–1044.
- 10.↑
Fine PE , LeDuc JW , 1978. Towards a quantitative understanding of the epidemiology of Keystone virus in the eastern United States. Am J Trop Med Hyg 27: 322–381.
- 11.↑
Henry CJ , Pillai AN , Morris JG , Hladish TJ , 2022. Ecology and public health burden of Keystone virus in Florida. Epidemics 39: 100555.
- 12.↑
Sudia WD , Newhouse VF , Calisher CH , Chamberlain RW , 1971. California group arboviruses: isolations from mosquitoes in North America. Mosq News 31: 576–600.
- 13.↑
Boyd KR , 1973. Studies on the Biologies of Mosquito Species Incriminated as Vectors of Keystone Virus in Houston, Texas. Texas University at Houston. Available at: https://apps.dtic.mil/sti/citations/ADA955160. Accessed November 17, 2021.
- 14.↑
Darsie RF Jr. , Ward RA , 2016. Identification and Geographical Distribution of the Mosquitoes of North America, North of Mexico. Gainesville, FL: University Press of Florida.
- 15.↑
Nzelu CO , Cáceres AG , Arrunátegui-Jiménez MJ , Lañas-Rosas MF , Yañez-Trujillano HH , Luna-Caipo DV , Holguín-Mauricci CE , Katakura K , Hashiguchi Y , Kato H , 2015. DNA barcoding for identification of sand fly species (Diptera: Psychodidae) from leishmaniasis-endemic areas of Peru. Acta Trop 145: 45–51.
- 16.↑
Folmer O , Black M , Hoeh W , Lutz R , Vrijenhoek R , 1994. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3: 294–299.
- 17.↑
Elbadry MA et al., 2021. Orthobunyaviruses in the Caribbean: Melao and Oropouche virus infections in school children in Haiti in 2014. PLoS Negl Trop Dis 15: e0009494.
- 18.↑
Thirion L , Pezzi L , Corcostegui I , Dubot-Pérès A , Falchi A , de Lamballerie X , Charrel RN , 2019. Development and evaluation of a duo chikungunya virus real-time RT-PCR assay targeting two regions within the genome. Viruses 11: E755.
- 19.↑
Pezzi L , Charrel RN , Ninove L , Nougairede A , Molle G , Coutard B , Durand G , Leparc-Goffart I , de Lamballerie X , Thirion L , 2020. Development and evaluation of a duo SARS-CoV-2 RT-qPCR assay combining two assays approved by the World Health Organization targeting the envelope and the RNA-eependant RNA polymerase (RdRp) coding regions. Viruses 12: E686.
- 20.↑
Thirion L , Dubot-Peres A , Pezzi L , Corcostegui I , Touinssi M , de Lamballerie X , Charrel RN , 2020. Lyophilized matrix containing ready-to-use primers and probe solution for standardization of real-time PCR and RT-qPCR diagnostics in virology. Viruses 12: 159.
- 21.↑
White SK , Mavian C , Salemi M , Morris JG Jr. , Elbadry MA , Okech BA , Lednicky JA , Dunford JC , 2018. A new “American” subgroup of African-lineage Chikungunya virus detected in and isolated from mosquitoes collected in Haiti, 2016. PLoS One 13: e0196857.
- 22.↑
Ahasan MS , Subramaniam K , Sayler KA , Loeb J , Popov V , Lednicky JA , Wisely SM , Krauer JMC , Waltzek T , 2019. Molecular characterization of a novel reassortment mammalian orthoreovirus type 2 isolated from a Florida white-tailed deer (Odocoileus virginianus). Fawn Virus Res 270: 197642.
- 23.↑
Nakamura T , Yamada KD , Tomii K , Katoh K , 2018. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34: 2490–2492.
- 24.↑
Larsson A , 2014. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30: 3276–3278.
- 25.↑
Bruen TC , Philippe H , Bryant D , 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 172: 2665–2681.
- 26.↑
Salemi M , Gray RR , Goodenow MM , 2008. An exploratory algorithm to identify intra-host recombinant viral sequences. Mol Phylogenet Evol 49: 618–628.
- 27.↑
Huson DH & Bryan D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23: 254–267.
- 28.↑
Martin DP , Murrell B , Golden M , Khoosal A , Muhire B , 2015. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1: vev003.
- 29.↑
Strimmer K , von Haeseler A , 2015. Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc Natl Acad Sci USA 94: 6815–6819.
- 30.↑
Nguyen L-T , Schmidt HA , von Haeseler A , Minh BQ , 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32: 268–274.
- 31.↑
Kumar S , Stecher G , Li M , Knyaz C , Tamura K , 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35: 1547–1549.
- 33.↑
Hamilton SB , Wyatt DE , Wahlgren BT , O'Dowd MK , Morrissey JM , Daniels DE , Lednicky JA , 2011. Higher titers of some H5N1 and recent human H1N1 and H3N2 influenza viruses in Mv1 Lu vs. MDCK cells. Virol J 8: 66.
- 34.↑
Grimstad PR , Calisher CH , Harroff RN , Wentworth BB , 1986. Jamestown Canyon virus (California serogroup) is the etiologic agent of widespread infection in Michigan humans. Am J Trop Med Hyg 35: 376–386.
- 35.↑
Lindsey NP , Lehman JA , Staples JE , Fischer M ; Division of Vector-Borne Diseases, National Center for Emerging and Zoonotic Infectious Diseases, CDC , 2014. West nile virus and other arboviral diseases – United States, 2013. Morb Mortal Wkly Rep 63: 521–526.